 |
Case Report
Twists in genetic mitochondria: Unraveling a case of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes overlapping with myoclonic epilepsy with ragged red fibers
1 OMS-III, Burrell College of Osteopathic Medicine, Las Cruces, NM, USA
Address correspondence to:
Jennifer H Phan OMS-III
NM 661-902-3778,
USA
Message to Corresponding Author
Article ID: 100076Z09JP2024
Access full text article on other devices

Access PDF of article on other devices


How to cite this article
Phan JH. Twists in genetic mitochondria: Unraveling a case of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes overlapping with myoclonic epilepsy with ragged red fibers. J Case Rep Images Med 2024;10(1):1–8.ABSTRACT
This case report presents a 23-year-old female with a complex medical history, initially diagnosed with juvenile myoclonic epilepsy, later discovered to have the m.3243A>G variant associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Despite a typical myoclonic epilepsy with ragged red fibers (MERRF) phenotype, genetic testing confirmed a singular MELAS mutation. The patient exhibited diverse symptoms, including seizures, chronic migraines, myoclonus, and visual disturbances. Diagnostic studies revealed basal ganglia calcifications and progressive brain MRI abnormalities consistent with mitochondrial dysfunction. Additionally, ophthalmologic exams identified features indicative of MELAS pigmentary retinopathy.
The discussion highlights the clinical and genetic diversity of mitochondrial disorders, emphasizing the challenges in distinguishing overlapping phenotypes. The patient’s mitochondrial DNA heteroplasmy influenced the variable onset and severity of symptoms. The case underscores the importance of comprehensive genetic investigations, as the m.3243A>G variant is associated with both MELAS and MERRF, leading to diagnostic complexities.
Management strategies, primarily symptom-focused due to the lack of a standardized treatment approach, include prophylactic arginine, taurine, and coenzyme Q10 supplementation. The patient’s response to various medications, including vagal nerve stimulator placement, Botox injections, and novel treatments like ASP0367, demonstrates the ongoing efforts to address symptoms and improve quality of life.
This report contributes to the understanding of mitochondrial overlap syndromes, offering insights into the diagnostic challenges and management complexities associated with MELAS and MERRF. The case underscores the need for a multidisciplinary approach, combining clinical, genetic, and therapeutic considerations, to optimize care for individuals with mitochondrial encephalopathies.
Keywords: Lactic acidosis, Migraine headaches, Myoclonic epilepsy, Ragged-red fibers
Introduction
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) is a maternally inherited disorder caused by mtDNA pathogenic variants, specifically the m.3243A>G variant, that affects multiple systems [1]. Symptomatic onset may be seen between the ages of 2 and 40 years with common clinical presentations other than those inherent in the name, including recurrent headaches, vomiting, muscle weakness, hearing impairment, etc. [1]. Individuals normally present with lactic acidosis, magnetic resonance imaging/computed tomography (MRI/CT) performed during stroke-like episodes typically will show increased signal areas that have no correspondence to the standard vascular distribution, and biopsy of the muscle typically show pathognomonic ragged red fibers [1]. Myoclonic epilepsy with ragged red fibers (MERRF) is also a maternally inherited disorder, most commonly caused by the m.8344A>G variant, with common manifestations of myoclonus and subsequent epilepsy, muscle weakness, ataxia, and findings of ragged red fibers [2]. Here, we present a patient with three years of convulsive seizures and the typical phenotype of MELAS and MERRF but was discovered with the mutation variant of only MELAS.
Case Report
A 23-year-old right-handed female presented to the emergency department with severe headache, nausea, vomiting, loss of consciousness, and witnessed convulsive seizures. Lumbar puncture was negative for the cause of her seizures. Shortly after hospitalization, she was diagnosed with juvenile myoclonic epilepsy and started on Levetiracetam 500 mg three times daily and Vimpat 200 mg twice daily.
The patient had a history of multi-focal myoclonic jerks since age 10. She had intractable migraine headaches with visual auras, about 2 times per week with 8–10 migraine days per month, which began in her early teens and has worsened since her early 20s.
Despite medication, she continued to experience occasional brief myoclonic jerks about 2–3 times a day; however, they were less severe. Her migraine headaches decreased in severity with seizure treatment. Her medications at the time included Levetiracetam 500 mg three times daily, Vimpat 200 mg bid, Nexplanon implant, albuterol inhaler as needed, and melatonin as needed. She was started on Verapamil 40 mg qhs for headaches and tachycardia, Sumatriptan 100 mg bid as needed for migraines, Aleve 200 mg bid as needed for headaches, and Promethazine 25 mg tid as needed for nausea.
At later follow-up appointments, Ajovy 225 mg was added for her migraines which improved over 50% (decrease from 10 days to 2 days per month). Vimpant was tapered off due to poor insurance coverage. However, the patient continued to have intractable epilepsy with myoclonic jerks multiple times daily.
Three year later, she was admitted to the hospital with intractable headaches and seizure activity with myoclonic jerks, nausea, and vomiting. She underwent extensive evaluation with noted abnormal brain magnetic resonance imaging (MRI) and elevated lactic acid and was subsequently diagnosed with MELAS/MERRF with cerebral edema and intractable headaches. Three months later, the patient proceeded with vagal nerve stimulator placement. Lamotrigine was stopped due to sedation and Nurtec ODT 75 mg qd was started for migraine. Her seizures and myoclonic jerks improved with VNS placement; however, she continued to have some myoclonic jerks multiple times daily in her legs and back.
One year later, the patient underwent genetic testing for possible MELAS/MERRF which confirmed the diagnosis of MELAS/MERRF due to 27% heteroplasmy (buccal) for the m.3243A>G variant in MT-TL1. Intrauterine device (IUD) birth control was recommended to avoid hormonal interactions and breakthrough ovulation while taking antiepileptic medications; however, the patient elected to undergo tubal ligation the following month.
Despite being on multiple medications, the patient continued to have chronic migraines with daily headaches about 3–4 times per week. She reported that Naratriptan, Aleve, and Promethazine helped with acute migraines, but her headaches still returned. Due to over 15 headache days per month, with headaches lasting over 4 hours, 155 units of botox injections were started and treatments have since continued every three months. The targeted muscles include the corrugator, procerus, frontalis, temporalis, occipitalis, cervical paraspinals, and trapezius. Her migraine headaches decreased with botox treatment, but they still returned so she was started on Ubrelvy 100 mg bid as needed for acute migraines. She reported prolonged visual auras of flashing, colored lights almost daily just before and after botox treatment which may possibly be visual migraine auras versus seizure auras.
Five months later, the patient was referred due to longstanding trouble falling and staying asleep. It is possible that her daytime sleepiness was a combination of nocturnal insomnia due to poor sleep hygiene, anxiety, possible sleep disordered breathing, and polypharmacy. Most of her seizure medications were sedating and could be leading to unwanted napping during the day which then led to fragmentation of sleep at night. She was started on a trial of hydroxyzine, advised to track her sleep pattern on a sleep diary and eliminate daytime napping.
She is currently enrolled in a study to assess the efficacy, safety, tolerability of ASP0367 and participants with primary mitochondrial myopathy (PMM).
Excluded medications of the study included Verapamil so it was discontinued and Metoprolol ER 50 mg qhs was started for migraine and tachycardia.
Since then, the patient has had recurrent hospitalization with seizures and nonepileptic seizures, three years after her initial presentation.
Other history
The patient has had a tonsillectomy and ear tube placements as a child. She was single with no children and was a college student. She was drinking about 1–2 cups of coffee daily and denied any history of tobacco or alcohol abuse. Her mother and sister had migraine headaches but there was no family history of seizure disorder.
Diagnostic studies
• Computed topography (CT) head without contrast:
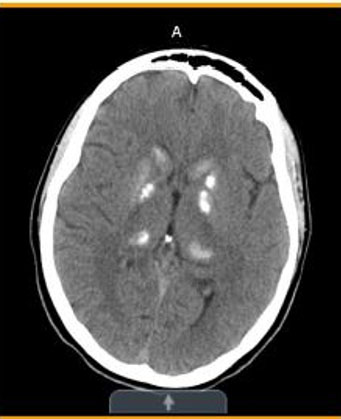
- Initial (Figure 1)—Stable mixed area calcifications/hyperdensities involving bilateral basal ganglia, globus pallidus, and thalami.
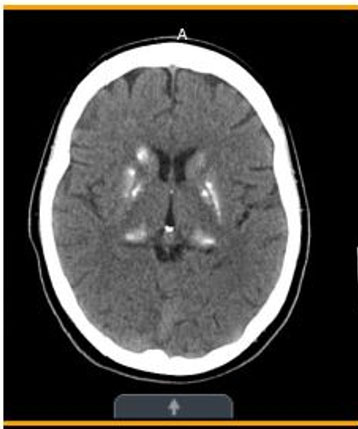
- Three years later (Figure 2)—Significant stable, heavy basal ganglia, and thalamus calcification.
• Magnetic resonance imaging (MRI) brain without contrast:
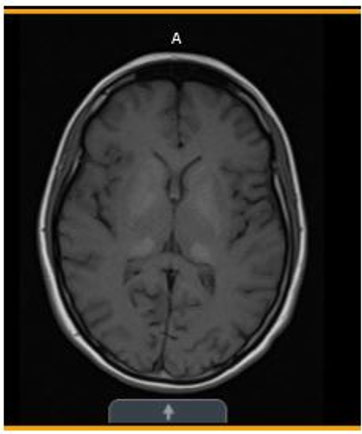
- Initial (Figure 3)—Subtle symmetric areas of T1 hyperintensity involving the bilateral basal ganglia/globus pallidi and thalami.
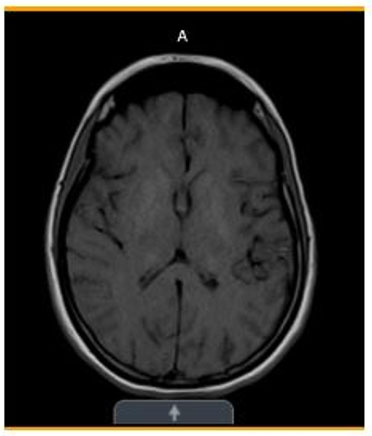
- One month later (Figure 4)—Stable symmetric bilateral abnormal signal in the basal ganglia and thalami with corresponding calcifications on the CT.
- Two months later (Figure 5)—Unchanged basal ganglia calcifications.
- One year later (Figure 6)—Moderate areas of restricted diffusion in the right occipital lobe, primarily with the cerebral cortex. Moderate brain edema: This combination of unilateral cerebral cortex restricted diffusion and basal ganglia calcification may be seen in patients with MELAS. Chronic findings of symmetrical, extensive, calcifications the bilateral basal ganglia and thalami.
- Two years later (Figure 7)—Normal MRI of the brain performed before and after intravenous (IV) contrast. Scans demonstrate no foci of abnormally increased T2 signal within the brain. No mass lesions are detected. No areas of abnormal enhancement are visible. No intraparenchymal or extra-axial hemorrhage is seen. No restricted diffusion is identified. None of the abnormal signal seen in the right occipital lobe on studies from previous 2 years are seen on the current exam. Presumably this was postictal ischemia that resolved.
- Two years, one month later (Figure 8)—Normal. None of the abnormal signals seen in the right occipital lobe on studies from previous two years are seen on the current exam. Presumably this was postictal ischemia that resolved.
- Three years later (Figure 9)—Similar finding of calcifications in bilateral basal ganglia, internal capsules, and dorsal thalami.
- Three years, one month later (Figure 10)—No acute intracranial abnormality.
Basal ganglia and thalamic calcifications consistent with MELAS
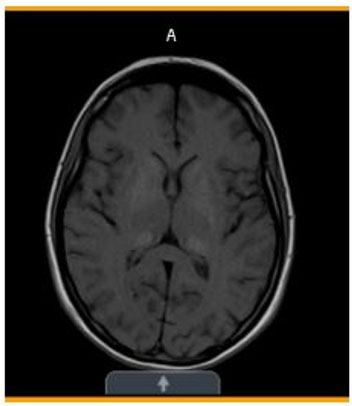
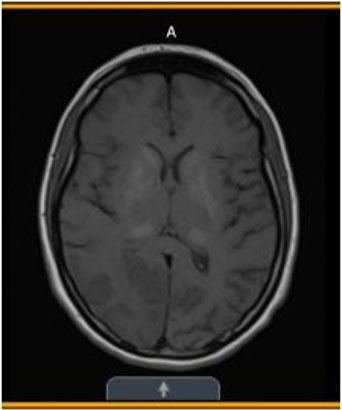
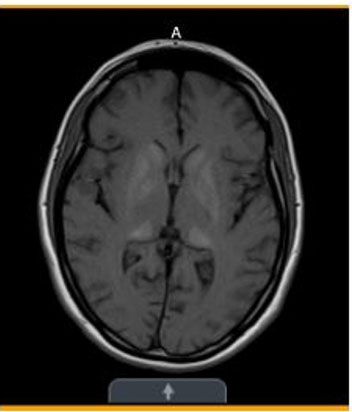
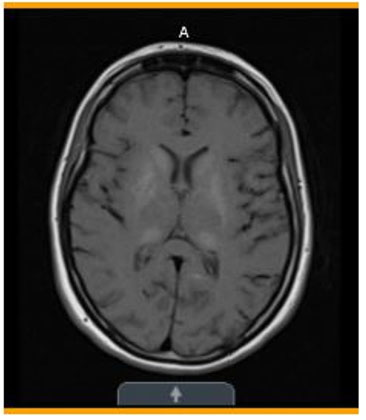
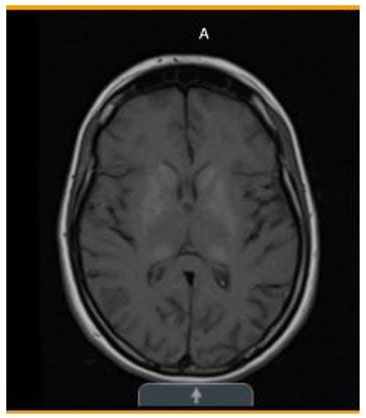
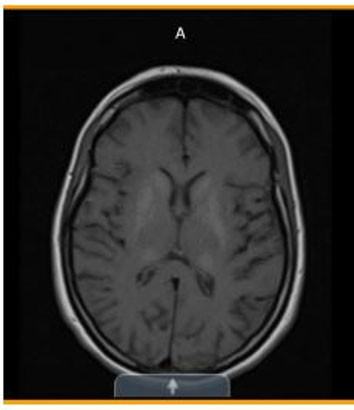
Diagnostic studies, continued...
- Sleep-deprived electroencephalogram (EEG) over 20–30 minutes:
- Initial—Mild diffuse background slowing is indicative of mild diffuse cerebral dysfunction but not specific as to etiology. 3 Hz spike-wave epileptiform discharges indicative of diffuse cortical irritability and could be consistent with a genetic generalized epilepsy. Photoparoxysmal response, one of which has associated myoclonus, is strongly suggestive of a genetic generalized epilepsy.
- One year later—Unchanged from prior. Abnormal due to frequent bursts of generalized atypical polyspike wave discharges seen throughout recording that was more prominent during drowsiness and light sleep. More prolonged bursts of epileptiform activity caused arousals from sleep. Findings suggest generalized epilepsy.
- Ophthalmologic examinations
- Optical coherence tomography (OCT): Subretinal deposits and retinal pigment epithelium (RPE) changes in bilateral eyes.
- Fundus: Pigment mottling showing macular pigment clumping circumferentially with white subretinal deposits. Retinal pigment epithelium (RPE) changes, loss of autofluorescence intensity. Changes consistent with MELAS pigmentary retinopathy.
Discussion
Mitochondrial overlap syndromes demonstrate both clinical and genetic diversity. Clinically, the patient fulfilled the criteria for MELAS and MERRF. Signs of MELAS phenotype include her longstanding history of seizures and episodes of focal neurological changes which resembled strokes. Although specific brain imaging features for MERRF have not been identified, this patient’s basal ganglia lesions matched the typical changes associated with this condition; cerebellar or brain stem changes have been reported in other cases but were not present in this patient [3]. The patient has a history of chronic migraines with visual aura, epilepsy with occipital seizures, myoclonus, and tachycardia, all of which are explained by her underlying diagnosis. She also endorses a history of chronic abdominal pain, vomiting, and poor appetite which are also common symptoms in MELAS/MERRF. She identified exercise as a trigger for her migraines; however, physical aerobic activity is also important for maintaining mitochondrial health/improving function. Findings of lactic acidosis prompted a muscle biopsy, which revealed presence of ragged red fibers and subsequently gave way to additional molecular diagnostic testing.
Diagnosing a patient with MELAS relies on fulfilling clinical diagnostic criteria and detecting a disease-causing variant in one of the MELAS-associated genes. In about 80% of MELAS cases, the pathogenic variant, m.3243A>G, is found in the mitochondrial gene MT-TL1 [1]. Approximately 80% of patients with MERRF syndrome carry the mitochondrial tRNA mutation, m.8344A>6 [4]. However, there has been rare instances where MERRF-like characteristics were documented in patients with MELAS variant, m.3243A>G [5], such as in the case of this patient.
Mitochondria are present within our cells throughout the body, with the purpose of creating cellular energy. Mitochondria are a major source of energy for our many organs and systems, such as the brain, eyes, hearing, heart, and muscles [6]. The mitochondria have their own DNA which codes for several genes involved in the maintenance and function of the mitochondria. This change is found in varying amounts as a percentage of heteroplasmy from person to person, family member to family member, and even among one person’s different tissues. Testing in blood or muscle, therefore, can provide helpful information, but does not by itself clearly indicate how clinically affected that person will be. In general, individuals with higher levels of heteroplasmy will have an earlier onset and more severe symptoms whereas those with lower levels of heteroplasmy have a later onset and less severe symptoms, and individuals with very low levels of heteroplasmy may be asymptomatic [7]. Generally, symptoms tend to be slowly progressive over time.
Inheriting MELAS/MERRF is maternal in nature as the mitochondria are passed onto offspring through the mother’s egg. Therefore, mothers who carry an mtDNA mutation have a higher risk of passing the DNA change on to each pregnancy. However, the amount passed along and resulting percent heteroplasmy is not predictable or controllable so an infant may have a lesser, similar, or higher percentage of heteroplasmy than the mother [7]. Carriers of an mtDNA mutation should be counseled on reproductive options and family planning including preimplantation genetic diagnosis, nuclear genome transfer, donor egg, prenatal testing, and adoption.
Regarding other family members, there is a significant chance that patients’ mothers will carry the m.3243 A>G variant and it is also highly likely, that any sisters, will have the 3243 variant, in varying percentages, in some of their cells [8]. Genetic counseling for siblings is recommended prior to testing for a more comprehensive discussion surrounding personal health risks and reproductive risks. As discussed above, mitochondria are only inherited from the oocyte, therefore, based on the percentage of heteroplasmy present in an egg cell, patients with MELAS/MERRF may be at risk of having a child with high heteroplasmy levels, who could be at risk of having more significant symptoms or earlier onset symptoms than the current patient has experienced. Prenatal diagnosis of mitochondrial gene changes in a fetus is technically possible; however, the interpretation of results is limited for most mutations.
The m.3243 A>G variant can be associated with variable clinical presentations, age of symptom onset, and disease course. The most common clinical presentation includes sensorineural hearing loss and diabetes, followed in frequency by stroke-like episodes, which the acronym, mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) has been historically used to describe [9]. Other findings reported in association with this variant include isolated myopathy, cardiomyopathy, neuropathy, seizures, migraine headaches, ataxia, psychiatric involvement, pigmentary retinopathy, cognitive impairment, gastrointestinal dysmotility, and short stature [10].
Phenotypic variability is influenced by various factors including tissue-specific heteroplasmy as well as other genetic and environmental effects. Heteroplasmy describes the mixture of normal and abnormal mtDNA in a cell. The percentage of heteroplasmy can vary between different tissue types in the same individual as well as between different affected individuals of the same family [11]. The higher the percentage of abnormal mtDNA, the greater mitochondrial dysfunction will occur in that cell. With some exceptions, the relative degree of heteroplasmy between tissue types is generally stable over the course of an individual’s lifetime; however, the level of heteroplasmy in certain tissue types such as the muscle may improve with exercise as new cells are generated [11].
Management
Currently, there is no widely accepted standardized treatment approach for individuals with MELAS/MERRF and the management primarily focuses on addressing the symptoms [12]. Initially, it is essential to conduct a thorough multidisciplinary assessment including neurological and cognitive assessments, neuroimaging, audiologic and ophthalmologic examinations, growth measurements, echocardiogram, electrocardiogram, and diabetic screenings to evaluate the involvement of multiple organs [12]. Obtaining an accurate diagnosis of a mitochondrial syndrome allows the treating physician to implement evidence-based treatment strategies and mitigate potential treatment- and disease-related complications such as seizures or febrile episodes. It is important to avoid medications such as valproic acid, phenobarbital, and tetracyclines due to their negative effects on the respiratory chain function [4].
In the event of an acute stroke-like episode, it is advisable to administer an intravenous arginine bolus within 3 hours of the onset of symptoms, followed by a continuous intravenous arginine infusion over the next 3–5 days [1]. For individuals who have experienced their first stroke-like episode, it is recommended to administer arginine prophylactically to decrease the recurrence of stroke-like episodes [12]. Daily maintenance prophylactic medications include arginine with taurine which has been shown to reduce the rate of stroke-like episodes in a few studies; however, the effects of taurine have not been studied by itself [13]. Arginine provides an extra substrate for nitric oxide production, promoting vasodilation while taurine helps with tRNA leucine modification, particularly beneficial in patients with an MT-TL1 mutation. These supplements collectively target various aspects of the underlying disease mechanism in 3243-related mitochondrial disease, setting it apart from other mitochondrial disorders.
Additionally, various supplementary co-factors have been used in the treatment of mitochondrial disease; however, the evidence in support of their efficacy is limited. Coenzyme Q10 (CoQ10), which aids in electron transfer and stabilizes the ETC complexes through its protective antioxidant effects, may yield positive results in improving muscle weakness, fatigability, and lactate levels in certain MELAS patients [12]. Creatine, being short-acting, can be helpful in the improvement of muscle strength and exercise tolerance [14]. Other supplements such as alpha-lipoic acid, levocarnitine, nicotinamide, and riboflavin are often administered to individuals with mitochondrial disease; however, their use in 3243-related mitochondrial disease is less well-documented [14].
Conclusion
This case report underscores the wide range of clinical manifestations seen in mitochondrial encephalopathies that begin in childhood. The patient described in this case contributes to the limited number of instances where mitochondrial m.3243A>G mutations are linked to clinical MERRF syndrome. It emphasizes the significance of conducting thorough genetic investigations when mitochondrial disease is suspected.
REFERENCES
1.
El-Hattab AW, Almannai M, Scaglia F. MELAS. In: Adam MP, Feldman J, Mirzaa GM, et al. editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2024.
[Pubmed]

2.
Huang G, Wang Y, Yao D. Myoclonic epilepsy with ragged red fibers syndrome associated with mitochondrial 3302A>G mutation in the MT-TL1 gene: A case report. Exp Ther Med 2023;25(2):87. [CrossRef]
[Pubmed]
3.
Ito S, Shirai W, Asahina M, Hattori T. Clinical and brain MR imaging features focusing on the brain stem and cerebellum in patients with myoclonic epilepsy with ragged-red fibers due to mitochondrial A8344G mutation. AJNR Am J Neuroradiol 2008;29(2):392–5. [CrossRef]
[Pubmed]
4.
Brackmann F, Abicht A, Ahting U, Schröder R, Trollmann R. Classical MERRF phenotype associated with mitochondrial tRNA(Leu) (m.3243A>G) mutation. Eur J Pediatr 2012;171(5):859–62. [CrossRef]
[Pubmed]
5.
Fabrizi GM, Cardaioli E, Grieco GS, et al. The A to G transition at nt 3243 of the mitochondrial tRNALeu(UUR) may cause an MERRF syndrome. J Neurol Neurosurg Psychiatry 1996;61(1):47–51. [CrossRef]
[Pubmed]
6.
Brand MD, Orr AL, Perevoshchikova IV, Quinlan CL. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br J Dermatol 2013;169 Suppl 2(0 2):1–8. [CrossRef]
[Pubmed]
7.
Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol 2013;5(11):a021220. [CrossRef]
[Pubmed]
8.
de Laat P, Janssen MCH, Alston CL, Taylor RW, Rodenburg RJT, Smeitink JAM. Three families with ‘de novo’ m.3243A > G mutation. BBA Clin 2016;6:19–24. [CrossRef]
[Pubmed]
9.
Orsucci D, Caldarazzo Ienco E, Rossi A, Siciliano G, Mancuso M. Mitochondrial syndromes revisited. J Clin Med 2021;10(6):1249. [CrossRef]
[Pubmed]
10.
Li D, Liang C, Zhang T, et al. Pathogenic mitochondrial DNA 3243A>G mutation: From genetics to phenotype. Front Genet 2022;13:951185. [CrossRef]
[Pubmed]
11.
Pickett SJ, Grady JP, Ng YS, et al. Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors. Ann Clin Transl Neurol 2018;5(3):333–45. [CrossRef]
[Pubmed]
12.
El-Hattab AW, Almannai M, Scaglia F. Arginine and citrulline for the treatment of MELAS syndrome. J Inborn Errors Metab Screen 2017;5:10.1177/2326409817697399. [CrossRef]
[Pubmed]
13.
Ohsawa Y, Hagiwara H, Nishimatsu SI, et al. Taurine supplementation for prevention of stroke-like episodes in MELAS: A multicentre, open-label, 52-week phase III trial. J Neurol Neurosurg Psychiatry 2019;90(5):529–36. [CrossRef]
[Pubmed]
14.
U.S. Department of Health and Human Services. (n.d.). Office of dietary supplements – dietary supplements for primary mitochondrial disorders. NIH Office of Dietary Supplements. [Available at: https://ods.od.nih.gov/factsheets/PrimaryMitochondrialDisorders-HealthProfessional/#ref]
SUPPORTING INFORMATION
Acknowledgments
Thank you to Dr. Timothy Ownbey, DO for his dedication to and patience with his students.
Author ContributionsJennifer H Phan OMS-III - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guaranter of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthor declares no conflict of interest.
Copyright© 2024 Jennifer H Phan, OMS-III. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.



